综述
Google学术搜索关键词 deep learning; genomics; review
云平台和软件资源 DeepLearning Resources
Applications of genomics:
- finding associations between genotype and phenotype
- discovering biomarkers for patient stratification
- predicting the function of genes
- charting biochemically active genomic regions such as transcriptional enhancers
Machine learning algorithms are designed to automatically detect patterns in data. A central issue is that classification performance depends heavily on the quality and the relevance of handcrafted features. Deep learning, a subdiscipline of machine learning, addresses this issue by embedding the computation of features into the machine learning model itself to yield end-to-end models
CNNs appplications:
- classifying transcription factor binding sites
- predicting molecular phenotypes such as chromatin features
- DNA contact maps
- DNA methylation
- gene expression
- translation effiency
- RBP binding
- microRNA (miRNA) targets
- predict the specificity of guide RNA
- denoise ChIP–seq
- enhance Hi-C data resolution
- predict the laboratory of origin from DNA sequences
- call genetic variants
- …
??? Modelling molecular phenotypes from the linear DNA sequence, albeit a crude approximation of the chromatin, can be improved by allowing for long-range dependencies and allowing the model to implicitly learn aspects of the 3D organization, such as promoter–enhancer looping.
RNNs applications:
- predicting single-cell DNA methylation states
- RBP binding
- transcription factor binding
- DNA accessibility
- miRNA biology …
GCNs applications:
- derive new features of proteins from protein–protein interaction networks
- modelling polypharmacy side effects
- predict various molecular properties including solubility, drug efficacy and photovoltaic efficiency
- predicting binarized gene expression given the expression of other genes
- classification of cancer subtypes
- …
??? In simple models such as linear models, the parameters of the model often measure the contribution of an input feature to prediction. Therefore, they can be directly interpreted in cases where the input features are relatively independent. By contrast, the parameters of a deep neural network are difficult to interpret because of their redundancy and nonlinear relationship with the output.
Feature importance scores can be divided into two main categories on the basis of whether they are computed using input perturbations or using backpropagation.
- Perturbation-based approaches systematically perturb the input features and observe the change in the output.
- Backpropagation-based approaches, to the contrary, are much more computationally efficient.
Advantages:
- end-to-end learning
- deal with multimodal data effectively
- abstraction of the mathematical details
基因组学历程:DNA双螺旋结构-1953,人类基因组计划-2001,基因组研究相关项目(FANTOM-2001,ENCODE-2013, Roadmap Epigenomics-2015等)
基因组学和传统遗传学:Genomic research aims to understand the genomes of different species. It studies the roles assumed by multiple genetic factors and the way they interact with the surrounding environment under different conditions. In contrast to genetics that deals with limited number of specific genes, genomics takes a global view that involves the entirety of genes possessed by an organism.
Models or strategies Researches Description CNN DeepBind, DeepSEA, Basset When applying convolutional neural networks in genomic, since deep learning models are always over-parameterized, simply changing the network depth would not account for much improvement of model performance. Researchers should pay more attention to particular techniques that can be used in CNNs, such as the kernel size, the number of feature map, the design of pooling or convolution kernels, the choice of window size of input DNA sequences, etc., or include prior genomic information if possible. RNN ProLanGO, DeepNano, DanQ, - Autoencoder - Now they have proved successful for feature extraction because of being able to learn a compact representation of input through the encode-decode procedure. When applying autoencoders, one should be aware that the better reconstruction accuracy does not necessarily lead to model improvement. CNN-RNN Deep GDashboard, - Transfer Learning & multitask learning PEDLA, TFImpute Transfer learning is such a framework that allows deep learning to adapt the previously-trained model to exploit a new but relevant problem more effectively Multi-view learning gRNM, Multi-view learning can be achieved by, for example, concatenating features, ensemble methods, or multi-modal learning. ??? Applications of deep learning in genomic problems have fully proven its power. Although the pragmatism of deep learning is surprisingly successful, this method suffers from lacking the physical transparency to be well interpreted so as to better assist the understanding of genomic problems.
Genomics applications:
- Gene Expression
1.1 Characterization
1.2 Prediction- Regulatory Genomics
2.1 Promoters and Enhancers
2.2 Splicing
2.3 Transcription Factors and RNA-binding Proteins- Functional Genomics
3.1 Mutations and Functional Activities
3.2 Subcellular Localization- Structural Genomics
4.1 Structural Classification of Proteins
4.2 Protein Secondary Structure
4.3 Protein Tertiary Structure and Quality Assessment
4.4 Contact Map
Major limitations of the DL models in the genomics area:
- Model interpretation (the black box)
- The curse of dimensionality
- Imbalanced classes. Transfer learning can provide a solution to tackle the class imbalanced problem.
- Heterogeneity of data
- Parameters and hyper-parameters tuning


Interpretability is defined as:
The ability to explain or to present in understandable terms to a human. – Doshi-Velez
The science of comprehending what a model did. – Gilpin
which is the first step toward explainability.A classification of common DNN interpretation approaches


Multilayer neural networks and backpropagation

文章阅读
[DeepSEA] [Basset]
[DeepFIGV]
[DeepAccess]
- 基因表达
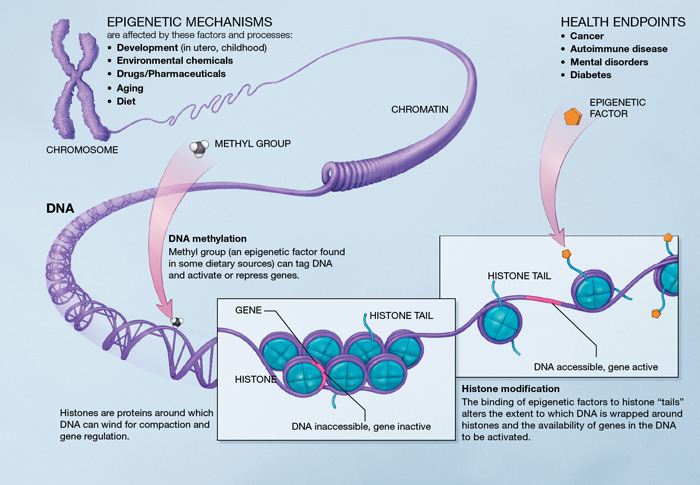
Histone modification levels are predictive for gene expression. 2010. /Rosa Karlić, Martin Vingron/
Drawbacks in the previous studies:
- they rely on multiple models to separate prediction and combinatorial analysis
- For input features, some of them take the average value of histone modification signal from the gene region and fail to capture the subtle differences among signal distributions of histone modifications
‘binning’ approach: that is, a large region surrounding the gene transcription start site (TSS) is converted into consecutive smaller bins.
Roadmap Epigenome Project, REMC
Model:


DeepDiff: DEEP-learning for predicting DIFFerential gene expression from histone modifications
Challenges:
- Genome-wide HM signals are spatially structured and may have long-range dependency.
- The core aim is to understand what the relevant HM factors are and how they work together to control differential expression.
- Since the fundamental goal of such analysis is to understand how HMs affect gene regulation, it requires the modeling techniques to provide a degree of interpretability and allowing for automatically discovering what features are essential for predictions.
- There exist a small number of genes exhibiting a significant change of gene expression (differential patterns) across two human cell types like A and B. This makes the prediction task using differential gene expression as outputs much harder than predicting gene expression directly in a single condition like A alone or B alone.
Input feature:
Model:

Deep learning decodes the principles of differential gene expression
Gene Expression Classification Based on Deep Learning
Gene expression inference with deep learning. 2016
- 转录因子
We developed Lisa to predict the transcriptional regulators (TRs) of differentially expressed or co-expressed gene sets. Based on the input gene sets, Lisa first uses histone mark ChIP-seq and chromatin accessibility profiles to construct a chromatin model related to the regulation of these genes. Using TR ChIP-seq peaks or imputed TR binding sites, Lisa probes the chromatin models using in silico deletion to find the most relevant TRs. Applied to gene sets derived from targeted TF perturbation experiments, Lisa boosted the performance of imputed TR cistromes and outperformed alternative methods in identifying the perturbed TRs.
Transcriptional regulators (TRs), which include transcription factors (TFs) and chromatin regulators (CRs), play essential roles in controlling normal biological processes and are frequently implicated in disease.
??? To infer the TRs that regulate a query gene set derived from differential or correlated gene expression analyses in humans or mice.
算法迭代:
MARGE builds a classifier based on H3K27ac ChIP-seq RPs from the Cistrome DB to discriminate the genes in a query differentially expressed gene set from a set of background genes.
BART extends MARGE, to predict the TRs that regulate the query gene set through an analysis of the predicted cis-regulatory elements.
Lisa (epigenetic Landscape In Silico deletion Analysis and the second descendent of MARGE), a more accurate method of integrating H3K27ac ChIP-seq and DNase-seq with TR ChIP-seq or imputed TR binding sites to predict the TRs that regulate a query gene set.
Changes in H3K27ac ChIP-seq and DNase-seq associated with cell state perturbations are often a matter of degree rather than switch-like. 细胞状态转变,涉及染色质状态的改变(组蛋白修饰 or 开放程度)只是程度改变而非有无的改变。

reading
可借鉴其研究思路和写作方式。
Prediction of cell type-specific, in vivo transcription factor binding sites is one of the central challenges in regulatory genomics. Here, we present our approach that earned a shared first rank in the “ENCODE-DREAM in vivo Transcription Factor Binding Site Prediction Challenge” in 2017. In post-challenge analyses, we benchmark the influence of different feature sets and find that chromatin accessibility and binding motifs are sufficient to yield state-of-the-art performance. Finally, we provide 682 lists of predicted peaks for a total of 31 transcription factors in 22 primary cell types and tissues and a user-friendly version of our approach, Catchitt, for download.
[DeepBind]
- 特征提取
DNA 序列特征: |分类|条目|描述| |–|–|–| |Physicochemical properties|Stacking energy| ||Enthalpy| ||Entropy| ||Flexibility_shift| ||Flexibility_slide| ||Free energy| ||Melting Temperature| ||Mobility to bend towards major groove| ||Mobility to bend towards minor groove| ||Probability contacting nucleosome core| ||Rise stiffness| ||Roll stiffness| ||Shift stiffness| ||Slide stiffness| ||Tilt stiffness| ||Twist stiffness| |Conformational properties|Bend| ||Rise| ||Roll| ||Inclination| ||Major Groove Depth| ||Major Groove Distance| ||Major Groove Size| ||Major Groove Width| ||Minor Groove Depth| ||Minor Groove Distance| ||Minor Groove Size| ||Minor Groove Width| ||Shift| ||Propeller Twist| ||Slide| ||Tilt| ||Tip| ||Twist| |Nucleotide content|Adenine content| ||Cytosine content| ||GC content| ||Guanine content| ||Keto (GT) content| ||Purine (AG) content| ||Thymine content| ||Pyrimidine (CT)|